BER200 - Problem 3
Ion Crystalization and Dynamics |
||||||||||||||||||||||||||||||||
Introduction: |
||||||||||||||||||||||||||||||||
|
The purpose of this exercise is to investigate the structure and
dynamics of so-called ions trapped in a electric and magnetic field
(Paul
Trap).
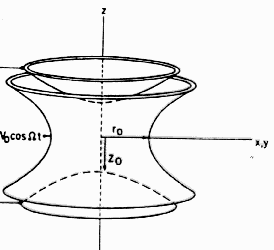
Ions in free space repel each other
due to Coulomb force repulsion and expand to infinity. In a trap
(Fig. below) an
additional attractive potential keeps the system bounded. For certain
parameters of the potential the most favorable energetic configuration
(the state with lowest energy) of
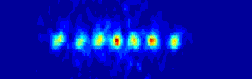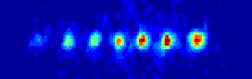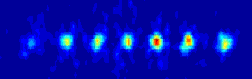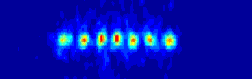
the ions will be to organize them self in a linear string.
Such a string is at present one of the most promising candidates for
implementing a quantum processor, which have the potential of solving
classical exponential problems in linear time. In this exercise we
will apply a classical model and simulation package to explore how
many atoms we are able to align before our string system changes structure
for a given set of experimental realistic trap parameters.
|
||||||||||||||||||||||||||||||||
Physical Problem Description: |
||||||||||||||||||||||||||||||||
|
On the atomic scale a set of particles is most accurately
described with
Schrödingers equation,
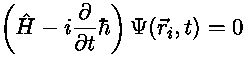
where
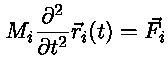 The forces can be calculated from the potential energy function
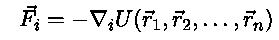 The potential is a sum of electrostatic repulsions and trap attractions,
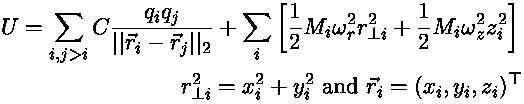
where
UnitsThe mass Mi is defined in [amu], qi, qj are defined in [e] and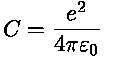 . .From experimentalists we learn that a typical trap parameter:
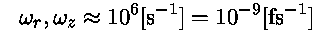
In the present exercise we apply a 10 times weaker
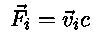
With this extra force each particle will be slowed down and the system
will move to equilibrium where all particles are stationary. Note
that too strong friction will freeze the system before reaching
equilibrium, and with too weak friction the system will oscillate
without converging to equilibrium. An additional check of whether
the final structure has reached equilibrium is provided by the fact
that for spherical system the Coulomb energy of the system is equal to
twice the trapping energy. |
||||||||||||||||||||||||||||||||
ProtoMol Quickstart: |
||||||||||||||||||||||||||||||||
|
ProtoMol ProtoMol is designed to be an important tool for Molecular Dynamics (MD) simulations. With a simple configuration setup, compatibility with other popular MD file formats, and the ability to run on parallel systems, ProtoMol combines high performance and ease of use. What ProtoMol provides is a generic, object-oriented component framework for MD simulations. To meet these high performance expectations, ProtoMol uses cell algorithms, grid techniques and well-optimized libraries to challenge the most computationally expensive forces. The design of ProtoMol also includes parallelization, based on components to distribute the work and data. The approach follows an incremental and partial parallelization scheme, which allows the developer to start with a sequential implementation and then do step by step parallelization. The overall framework of ProtoMol is designed for non-bonded, bonded, short-range and long-range forces for systems with tens of thousands of atoms representing water and several large molecules. It is designed for high flexibility, ease in extension and maintainence, and high performance demands. MD describes a molecular system as a function of time based on the integration of Newton's equations of motion and interacting forces. The integrators are the part of the program that solve the differential equations that describe the system. Specifically, the integrators provide a set of forces, that describe the system at each time. Thus, it is easy to see that the central part of the entire system is the integrators with their force definitions. ProtoMol provides several integrators (including multiple-time-stepping) and a variety of forces (fast approximations and exact algorithms) to describe and solve Newton's equations of motion or more advanced MD equations. The definition of integrators and forces are part of the configuration file, such that there is no need of re-compilation to change the integrator scheme or the set of forces. All this together makes ProtoMol a favorable MD framework to experiment with different integrator schemes and/ or different force(-algorithms)/ potentials in a sequential or parallel environment. Download - Binaries and Source: Release 1.8.3 Simple Test Case I: 2-Type/Shell 3Mg+24 7Ca+40 (Leapfrog and friction)
Simple Test Case II: 1-Type/Shell 20,288Ca+40 (Nosé-Hoover and Multigrid)
Running ProtoMol
./protomol 3mg7ca.confIn order to get help: ./protomol -hTo figure out the supported forces: ./protomol -fTo figure out all possible keywords: ./protomol -kVisualization ProtoMol writes DCD files, which can be read by VMD or XYZ trajectory files visualized by the OpenGL/GLUT viewer (xyzviz.cpp). ProtoMol Units
 1 Velocities in PDB files are scaled to get a more accurate representation since the PDB format has a limited representation of floating numbers. More Information
|
||||||||||||||||||||||||||||||||
Problem 1: |
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
Problem 2: |
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
Problem 3: |
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
Related Links: |
||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
Report: |
||||||||||||||||||||||||||||||||
The report should be published on the web and must include:
|
||||||||||||||||||||||||||||||||
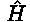 is the Hamiltonian of the ions interacting with the
time dependent electro magnetic fields and
is the Hamiltonian of the ions interacting with the
time dependent electro magnetic fields and 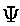 is the wave function. This problem is extremely
difficult for more than 2 particles but fortunately for sufficient
massive particles (like ions) at sufficient high temperatures, we
may skip all quantum effects and attack it classically. In classical
mechanics the dynamics of the particles is determined by Newtons 2. law,
is the wave function. This problem is extremely
difficult for more than 2 particles but fortunately for sufficient
massive particles (like ions) at sufficient high temperatures, we
may skip all quantum effects and attack it classically. In classical
mechanics the dynamics of the particles is determined by Newtons 2. law,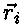 is the coordinate of
particle i.
is the coordinate of
particle i.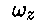 .
To obtain a stable configuration the experimentalists apply lasers in the
z-direction which can cause the ions to slow down (
.
To obtain a stable configuration the experimentalists apply lasers in the
z-direction which can cause the ions to slow down (


{kind=link}